SMA is a rare genetic neuromuscular disease that affects the part of the nervous system that controls voluntary muscle movement
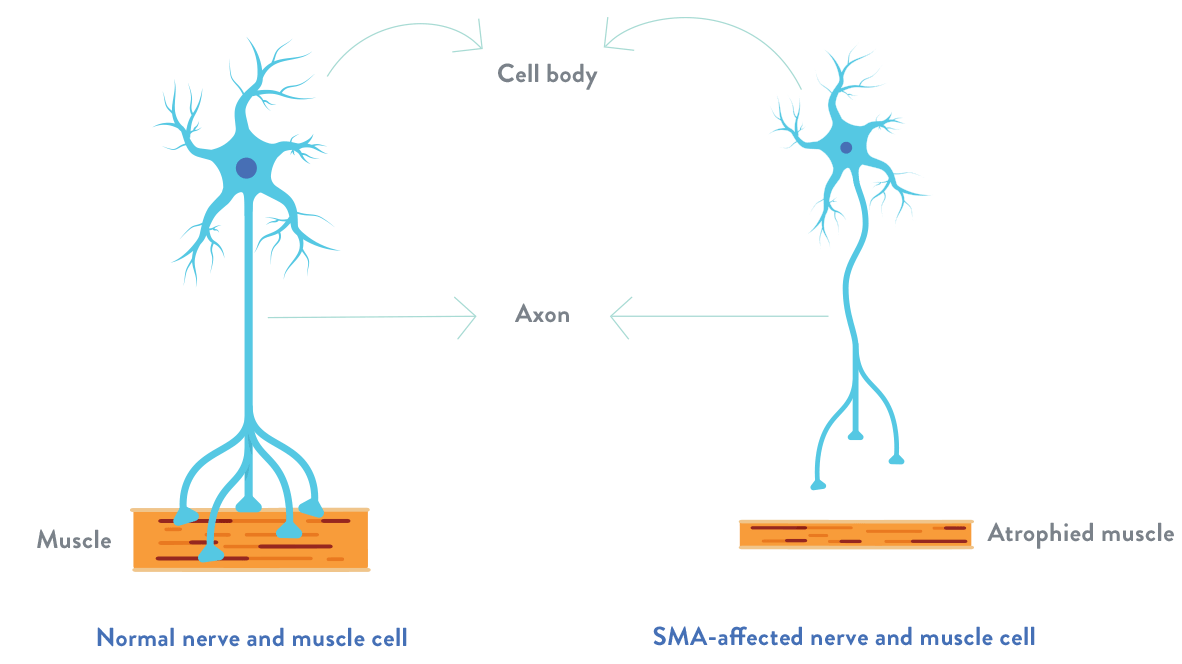
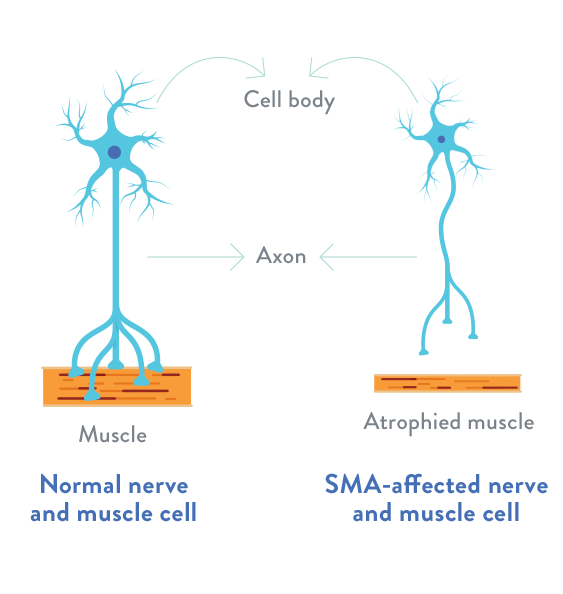
In spinal muscular atrophy, there is a loss of important cells in the spinal cord called motor neurons, which are essential for muscle strength and movement. These motor neurons regulate muscle activity by sending signals from the central nervous system (CNS), which is the part of the body’s nervous system that includes the brain and spinal cord.
The loss of functioning motor neurons leads to progressive muscle weakness and atrophy (the gradual decrease in the mass and strength of muscles), as muscles stop receiving signals from the CNS.
Unlike many other rare neuromuscular diseases, there is a clear understanding of the specific genetic cause of spinal muscular atrophy.
What causes spinal muscular atrophy?
Spinal muscular atrophy is caused by a mutation in the survival motor neuron 1(SMN1) gene. This gene is responsible for producing survival motor neuron (SMN) protein, which maintains the health and normal function of motor neurons. In individuals with spinal muscular atrophy, both copies of the SMN1 gene are mutated, leading to decreased production of SMN protein. Without a proper level of SMN protein, motor neurons in the spinal cord will be lost, preventing the muscles from receiving proper signals from the brain.
The degeneration of motor neurons leads to the gradual decrease in the mass and strength of muscles (atrophy).
For illustrative purposes only.
What are the effects of spinal muscular atrophy?
Spinal muscular atrophy affects everyone differently, and it is important to note that symptoms can vary greatly according to the age of onset and disease severity. Individuals may experience progressive muscle weakness in the muscles closest to the center of the body, such as the shoulders, thighs, and pelvis. These muscles enable activities such as head control/movement, sitting up, crawling, and walking. Breathing and swallowing may also be affected.
It is believed that spinal muscular atrophy does not affect the neurons responsible for cognition, which is the mental process through which we gain knowledge and understanding through thought, experience, and the senses. According to one study, children and adolescents with spinal muscular atrophy have normal intelligence, with IQs in the standard range. For school-age children, special accommodations may be needed to help maintain cognitive and intellectual engagement.
What should I know about SMN2?
All individuals with spinal muscular atrophy have at least one “backup gene,” known as SMN2. The SMN2 gene has a similar structure to SMN1, but only a small amount (10%) of the SMN protein it produces is fully functional. This low level of SMN protein is not effective enough to sustain the survival of motor neurons in the CNS.
The number of SMN2 genes may vary, and a higher SMN2 copy number is associated with less-severe symptoms of spinal muscular atrophy. Nevertheless, the disease has a wide range of symptoms and it is difficult to predict severity based on the number of SMN2 copies alone. Thus, experts recommend that care decisions be made based on the individual’s functional ability and not on SMN2 copy number alone.
The discovery of this backup gene provides a unique opportunity for the development of potential therapies that may help the SMN2 gene produce more SMN protein.
Is spinal muscular atrophy genetic?
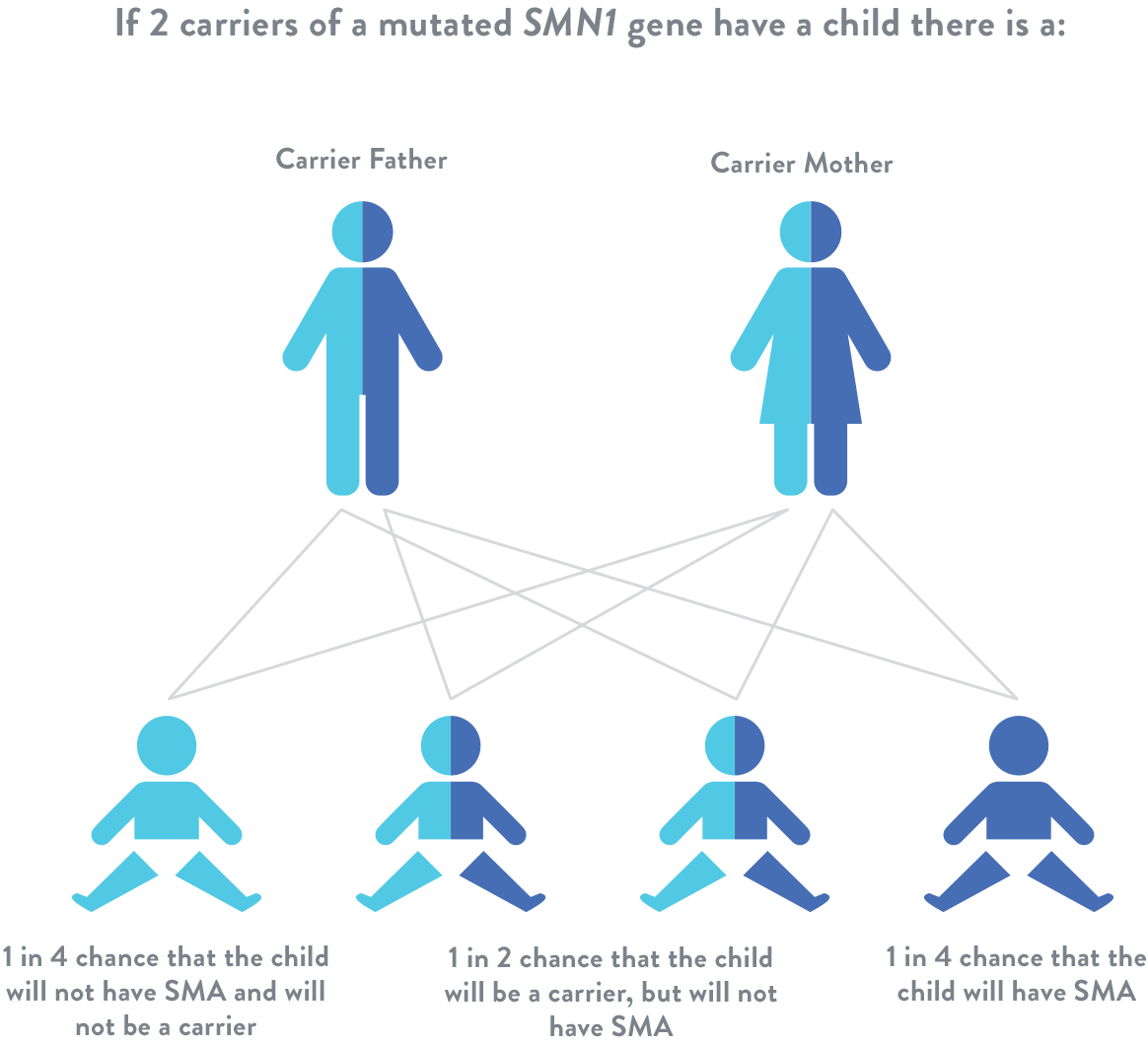
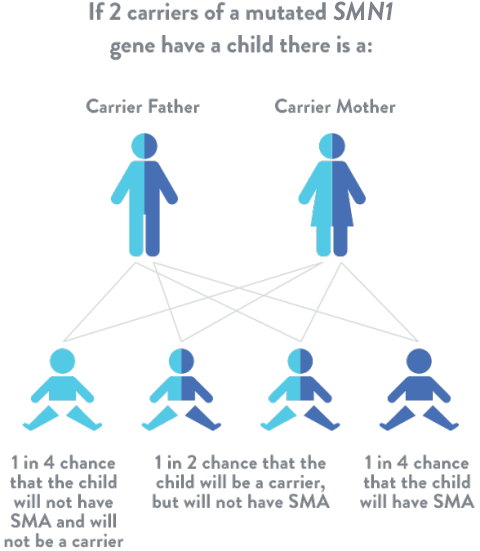
Spinal muscular atrophy is an autosomal recessive disease, which means that for a individual to be at risk, he or she must inherit 1 mutated SMN1 gene from each parent. If a child inherits only 1 mutated SMN1 gene, they are considered a “carrier,” but usually do not have symptoms of spinal muscular atrophy.
SMA carrier testing
If you have a family history of spinal muscular atrophy, your chances of being a carrier are greater than average. In making reproductive decisions, it may be helpful to consult with your physician and/or genetic counselor to learn what mutation(s) are common in your family. Once your family mutation(s) are known, an appropriate genetic testfor your situation may be determined:
If your family mutation(s) are SMN1 deletions, then copy number testing may be appropriate for you
If your family mutation(s) include a more subtle change in the gene, then your physician and the laboratory may decide whether testing can be done to look for that specific change
If you are unable to obtain your family mutation(s) information, you can still have a copy number test performed. Your chance of being a carrier (before you have testing) will be calculated from your family history. If your results are normal, your chance of being a carrier may be lower.
Many laboratories and hospitals offer spinal muscular atrophy carrier screening to determine whether 1 or both parents are carriers of the mutated SMN1 gene. This can provide individuals and families with information about the risk of giving birth to a child with spinal muscular atrophy. A genetic counselor is trained to make information about genetic risks, testing, and diagnosis easier for families to understand.